
How much documentation does my Software as a Medical Device regulatory submission really need?
A challenge that Software as a Medical Device and AI developers often face is to define the quantity and content of documentation that is required for the regulatory certification (CE marking or FDA clearance) of their product. Here, we explain how to derive the documentation requirements from the safety and risk class of the device and provide a free cheat sheet to help you navigate regulatory submissions.
The Agile Manifesto dilemma
Many software developers new to medical devices are proponents of the Agile Manifesto. This contains four value statements:
“Individuals and interactions over processes and tools”
“Working software over comprehensive documentation”
“Customer collaboration over contract negotiation”
“Responding to change over following a plan”
The Agile Manifesto goes on to state that “while there is value in the items to the right (of each sentence), we value the items on the left more”. The problem when it comes to SaMD regulators is that they actually want to see evidence of the things on the right - the processes, the documentation, the plan.
So, agile developers coming into the SaMD sector need to reframe that last statement as follows: “While we see the importance of the items on the left, we must honour the items on the right in order to provide objective evidence to the regulators and others that our Software as a Medical Device is safe, effective and cybersecure”.
This isn’t just our view: the American Association for the Advancement of Medical Instrumentation (AAMI) has published guidance on the use of AGILE practices in the development of medical device software, the latest edition being AAMI TIR 45:2023.
Documentation (evidence) is necessary to demonstrate compliance to regulations and to facilitate the investigation into software problems, as well as to evaluate software for those devices requiring regulatory approval, e.g., approval, clearance, licensing, registration, self-certification, etc.
Regulatory documents often do not get into the details of how these practices must be performed, leaving those details up to the manufacturer to define. This approach allows manufacturers to create their own practices that align with regulatory principles.
This article offers a way to create practices that align both with regulatory principles and with an agile mindset.
SaMD and IEC 62304
For the UK, the EU and the rest of the world we have the standard BS EN 62304:2006+A1:2015 (originally published by the International Electrotechnical Commission (IEC) as IEC 62304) which defines software development lifecycle (SDLC) processes, including software safety classification – the safety classification of the software drives the content of documentation. We use 62304 as part of claiming the presumption of conformity to the medical device regulatory rules for the UK and for the EU - you can read more about IEC 62304 in our other blog.
For the USA, an additional layer is required – documentation level evaluation – as defined in the FDA’s Content of Premarket Submissions for Medical Device Functions guidance document issued in June 2023. This guidance replaces older guidance, Content of Premarket Submissions for Software Contained in Medical Devices, that dates back to 2005 – this older guidance introduced the concept of Level of Concern that may still be familiar to some, but which has been deprecated and replaced by Document Level Evaluation
The point is that the level of SaMD documentation that is required is aligned with the risk classification of the software.
Software as a Medical Device safety classification and document level evaluation
To know what quantity of documentation you need, you first must understand your software safety classification. Your documentation is then subsequently designed to demonstrate that your software medical device is safe, effective and cyber-secure, given the level of risk it poses to the patient, to other users or the environment.
| 62304 §4.3 | FDA premarket guidance §5 |
|
The manufacturer shall assign to each SOFTWARE SYSTEM a software safety class (A, B, or C) according to the RISK of HARM to the patient, operator, or other people resulting from a HAZARDOUS SITUATION to which the SOFTWARE SYSTEM can contribute in a worst-case scenario… The SOFTWARE SYSTEM is software safety class A if: – the SOFTWARE SYSTEM cannot contribute to a HAZARDOUS SITUATION; or – the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which does not result in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM. The SOFTWARE SYSTEM is software safety class B if: – the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which results in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM and the resulting possible HARM is non-SERIOUS INJURY. The SOFTWARE SYSTEM is software safety class C if: – the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which results in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM and the resulting possible HARM is death or SERIOUS INJURY. NOTE 1: External RISK CONTROL measures can be hardware, an independent SOFTWARE SYSTEM, health care procedure, or other means to minimize that software can contribute to a HAZARDOUS SITUATION. |
Basic Documentation should be provided for any premarket submission that includes device software function(s) where Enhanced Documentation does not apply. Enhanced Documentation should be provided for any premarket submission that includes device software function(s) where a failure or flaw of any device software function(s) could present a hazardous situation with a probable risk of death or serious injury, either to a patient, user of the device, or others in the environment of use. These risks should be assessed prior to implementation of risk control measures. Sponsors should consider the risks in the context of the device’s intended use (e.g., impacts to safety, treatment, and/or diagnosis), and other relevant considerations. Serious Injury- An injury or illness that: 1) Is life-threatening, 2) Results in permanent impairment of a body function or permanent damage to a body structure, or 3) Necessitates medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure. Permanent is defined as irreversible impairment or damage to a body structure or function, excluding trivial impairment or damage. |
We can see that the 62304 Safety Classes A + B roughly correspond to the FDA’s Basic Documentation requirement, whereas Safety Class C roughly corresponds to the Enhanced Documentation requirement.
Defining a Software System
Annex B, section B.3, of 62304 describes a SOFTWARE SYSTEM thus:
This standard chose to use three terms to describe the decomposition of a SOFTWARE SYSTEM (top-level). The SOFTWARE SYSTEM can be a subsystem of a medical device (see IEC 60601-1-4) or a MEDICAL DEVICE in its own right, which then becomes a software MEDICAL DEVICE.
Section 3.35 of 62304 defines a HAZARDOUS SITUATION thus:
Circumstances in which people, property of the environment are exposed to HAZARDS
For the sake of this discussion, there are two types of medical device software system:
Software in a Medical Device (SiMD) – software embedded in a device with a fixed purpose, e.g. the software in a digital thermometer – this is what is meant above by “a subsystem f a medical device”
Software as a Medical Device (SaMD) – that is software running on a general computing platform e.g. cloud server, or even in a smartphone or smartwatch (in that other apps can run on that platform in parallel with the SaMD) – this is what is meant above by “a MEDICAL DEVICE in its own right”
Defining a Hazardous Situation
Turning to the concept of a hazardous situation, let’s use the example given in PD IEC/TR 80002-1:2019, the IEC’s guide on how to apply the international standard for medical device risk management to medical device software. Table A1 in this publication has a neat summary of how to look at software hazardous situations, in the context of medical device risk assessment, which I put here with a few changes of my own to make it more pertinent to triage/ detection/ diagnostic software; also to add dimensions of cybersecurity and Artificial Intelligence/ Machine Learning that are not anticipated in this publication:
Dimension |
HAZARD | Foreseeable sequence of events involving software | Initiating cause | HAZARDOUS SITUATION | HARM |
Patient safety | Electrical energy | Software output controlling laser power applied to an optical coherence tomography (OCT) retinal scan is too high | 1. Software algorithm limitations 2. Software is correctly specified but has an ANOMALY | Excessive laser power applied to the patient’s eye |
Burns Loss of vision |
Loss of clinical function |
Software fails to provide diagnostic function for which it is designed | 1. Software is unable to handle unusual input data 2. Software fails to detect incorrect setup of equipment 3. Hardware does not provide sufficient resources to support the timely operation of software> |
1. Device cannot provide triage/ detection/ diagnosis when needed 2. Device operates when not correctly setup 3. Device fails to warn of injurious or life-threatening condition | Deterioration of patient’s condition
Death | |
Neglect of patient by clinical staff | Software inputs and outputs confuse of mislead the user | 1. Software-human interface confuses the user 2. Software outputs exceed the user’s capacity to respond 3. User does not understand the software’s limitations | 1. Mistreatment of patient
2. Lack of response to emergencies 3. Over-reliance on software replaces personal initiatives (“automation bias”) | ||
Cyber-security | Loss of data integrity | See Table F.1 in PD CEN ISO/TR 24971:2020, the guide to the general application of the medical device risk management standard ISO 14971 | |||
Loss of data availability | |||||
Loss of data confidentiality | |||||
AI/ML | Data quality: incomplete data | See Table B.3 in BS/AAMI 34971:2023, the guide on the application of medical device risk management to machine learning in artificial intelligence | |||
Overfitting | |||||
Bias: proxy variables | |||||
Data storage: malicious ML | |||||
Overtrust: overconfidence | |||||
Mapping Software Safety Classes
Now that we have an idea of hazardous situations that are related to SaMD (and SiMD), and the possible consequent harms, we can have a go at mapping device risk classes for SaMD, using the EU risk classifications (I, IIa, IIb, III), against the Software Safety Classes tabulated above; also the FDA document level against the corresponding Software Safety Class:
|
SaMD risk class based on EU MDR Rule 11 |
62304 §4.3 |
FDA |
|---|---|---|
|
Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, |
The SOFTWARE SYSTEM is software safety class B if: – the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which results in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM and the resulting possible HARM is non-SERIOUS INJURY. |
Basic |
|
except if such decisions have an impact that may cause:
|
The SOFTWARE SYSTEM is software safety class C if: – the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which results in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM and the resulting possible HARM is death or SERIOUS INJURY. |
Enhanced |
|
The SOFTWARE SYSTEM is software safety class B if: – the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which results in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM and the resulting possible HARM is non-SERIOUS INJURY. |
Basic |
|
All other software is classified as class I. |
The SOFTWARE SYSTEM is software safety class A if: – the SOFTWARE SYSTEM cannot contribute to a HAZARDOUS SITUATION; or – the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which does not result in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM. |
One last, but supremely important, point about classification before we finally address the documentation that goes with each class: looking at the above, you could argue that “well, I’ve done my software risk assessment and evaluation, I’ve introduced risk control measures external to the software system, there cannot be any more unacceptable risk, so all my SaMD is in Safety Class A”. That argument, unfortunately, does not pan out. The point missed in that argument is that software can be buggy – even with the best SDLC process and the finest engineering team in the world software can still expose one or more of what 62304 defines as an ANOMALY, that is “any condition that deviates from the expected based on requirements specifications, design documents, standards, etc. or from someone’s perceptions or experiences. ANOMALIES may be found during, but not limited to, the review, test, analysis, compilation, or use of SOFTWARE PRODUCTS or applicable documentation”. So whilst we’ve seen, above, the definition of Software Safety Classes taken from 62304 section 4.2, we need to scroll all the way down to 62304 section 7.1 to get to these points:
§7.1.1
… NOTE: The hazardous situation could be the direct result of software failure or the result of the failure of a RISK CONTROL measure that is implemented in software.
§7.2.2
… the MANUFACTURER shall consider potential causes including, as appropriate:
1. incorrect or incomplete specification of functionality;
2. software defect in the identified SOFTWARE ITEM functionality;
3. failure or unexpected results from SOUP;
4. hardware failures or other software defects that could result in unpredictable software operation; and
5. reasonably foreseeable misuse.
Even though section 7.1 is written for Safety Class B + C software, we need to take note of the point in section 4.2g, that “For each SOFTWARE SYSTEM, until a software safety class is assigned, Class C requirements shall apply”.
So what does that all tell us?
We need to start off our risk analysis as if the software is in Safety Class C.
We can downgrade software items in our software system to Safety Class B or A depending on the outcome of the risk analysis
Per section 7.2.2 the downgrade of the Safety Class relies not only on the implementation of effective risk control measures but also on the harm that could be caused by residual bugs (“anomalies”).
Here is how this supremely important point is nicely summarised in PD IEC/TR 80002-1:2019 section 6.2.2.6:
It is accepted that increasing the rigour of the software development PROCESS can reduce the number of software ANOMALIES. It should be noted that while testing on MEDICAL DEVICE SOFTWARE can reduce the number of software ANOMALIES, it cannot be assumed that when the software passes all planned tests, no software ANOMALIES remain. This is because in clinical use the inputs to the software will include sequences that were not part of the planned tests.
Since MEDICAL DEVICE SOFTWARE is too complex to test exhaustively, rigorous testing can only be viewed as a method to lower the probability of HAZARDOUS SITUATIONS. Testing by itself, however, is not sufficient to establish confidence that the software can be treated as a high-integrity component.
So, the hazardous situations resulting from software cannot be eliminated, only the probability of hazardous situations occurring can be reduced. This is why not all software can be in Safety Class A.
SaMD documentation requirements cheat sheet
Now that we understand the Software Safety Classification and the FDA documentation level evaluation, to answer the question of just how much documentation is needed for a Software as a Medical Device regulatory submission, we can tabulate the following cheat sheet:
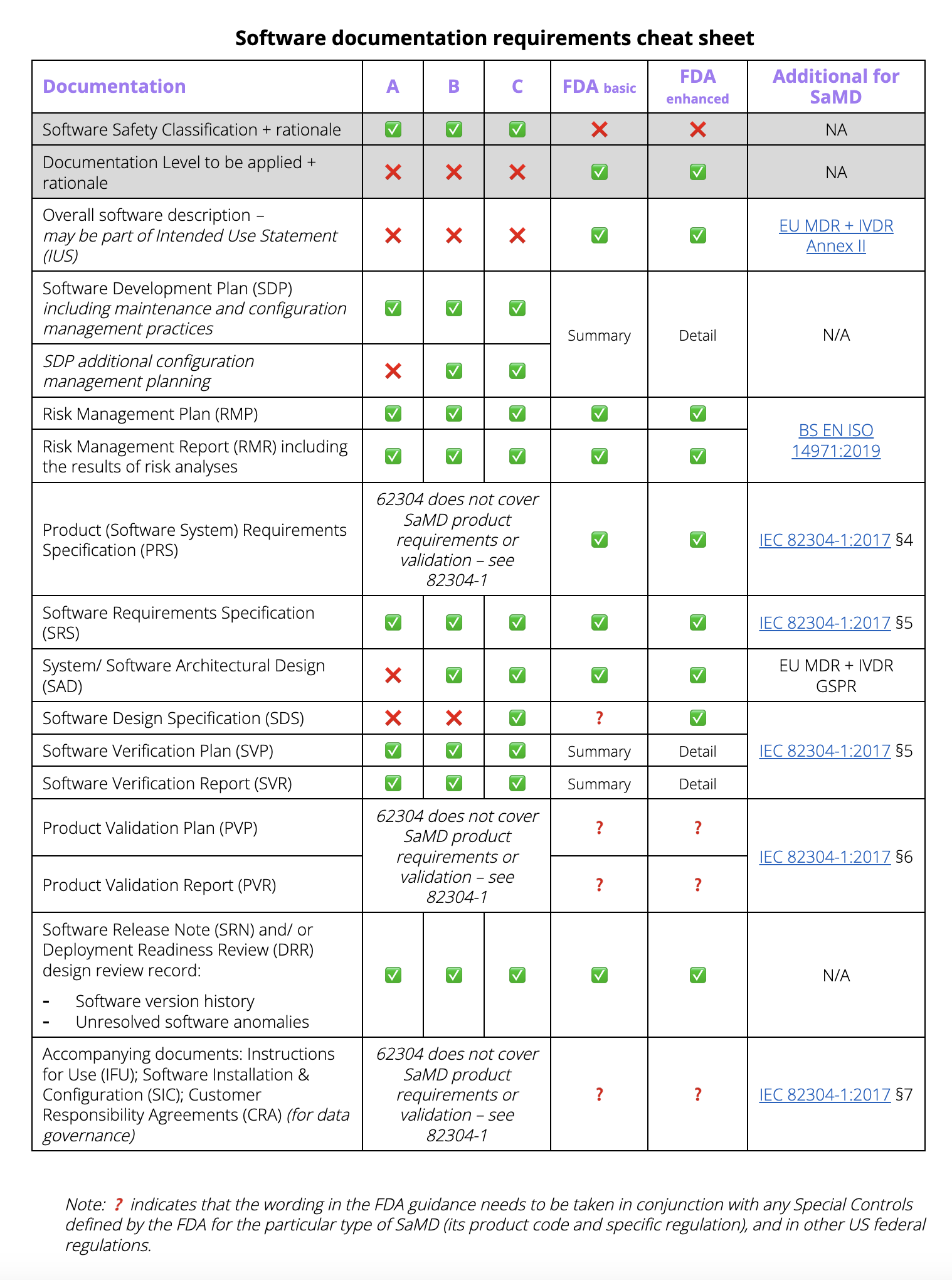
the SaMD documentation requirements cheat sheet
Not shown in the cheat sheet are cybersecurity-specific artefacts, which are called for by different standards and FDA guidance documents; ditto design documentation for AI/ML, covering for instance the Good Machine Learning Practice (GMLP) and Predetermined Change Control planning as additions to the SAD, SDS, SDP and RMP. So even where the SDS is not strictly speaking required for Safety Class B, some design documentation exposing GMLP is advisable. Finally, be aware that the FDA requires extra Special Controls for some products, even if their Basic level does not call for it.
At Hardian Health we can support you through risk classification, hazard identification and risk analysis, and the other aspects of your regulatory pathway for AI and software as a medical device.
Hardian Health is a clinical digital consultancy focused on leveraging technology into healthcare markets through clinical strategy, scientific validation, regulation, health economics and intellectual property.