
Medical Device File (MDF) Design Support
Your MDF provides evidence to demonstrate compliance of the device to all the applicable regulations. Its structure and design is key.
Your MDF (also known as Technical Documentation) is a collation of all the necessary regulatory, clinical and technical documents - starting from your Intended Use, all the way to Post Market Surveillance Reporting for the entire product lifetime.
When it comes to building medical software, you as a medical device manufacturer must submit MDF documentation to the authority before placing it on the market. MDF documentation is mandatory to prove that your device meets all applicable requirements as this justify the conformity assessment and certification approval process.
Writing concise, comprehensive and well-thought-out MDF documents:
Can help your internal staff by providing a central information source regarding your medical device.
Can reduce the review time spent by Regulatory Bodies (Notified bodies/Approved Bodies).
Makes the job of auditors significantly easier when it comes to looking for the information they need to assess whether your software meets the legal requirements.
Best Practices
Align with internationally recognised standards
The need for an MDF is mentioned in section 4.2.3 of ISO 13485:2016, 21 CFR 820.181 (referred to as Device Master Records) and in all the applicable regulations such as the EU Medical Device Regulations (EU MDR), the EU in vitro Diagnostic Medical Device Regulations (2017/746 EU IVDR) and the UK Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (UK MDR 2002).
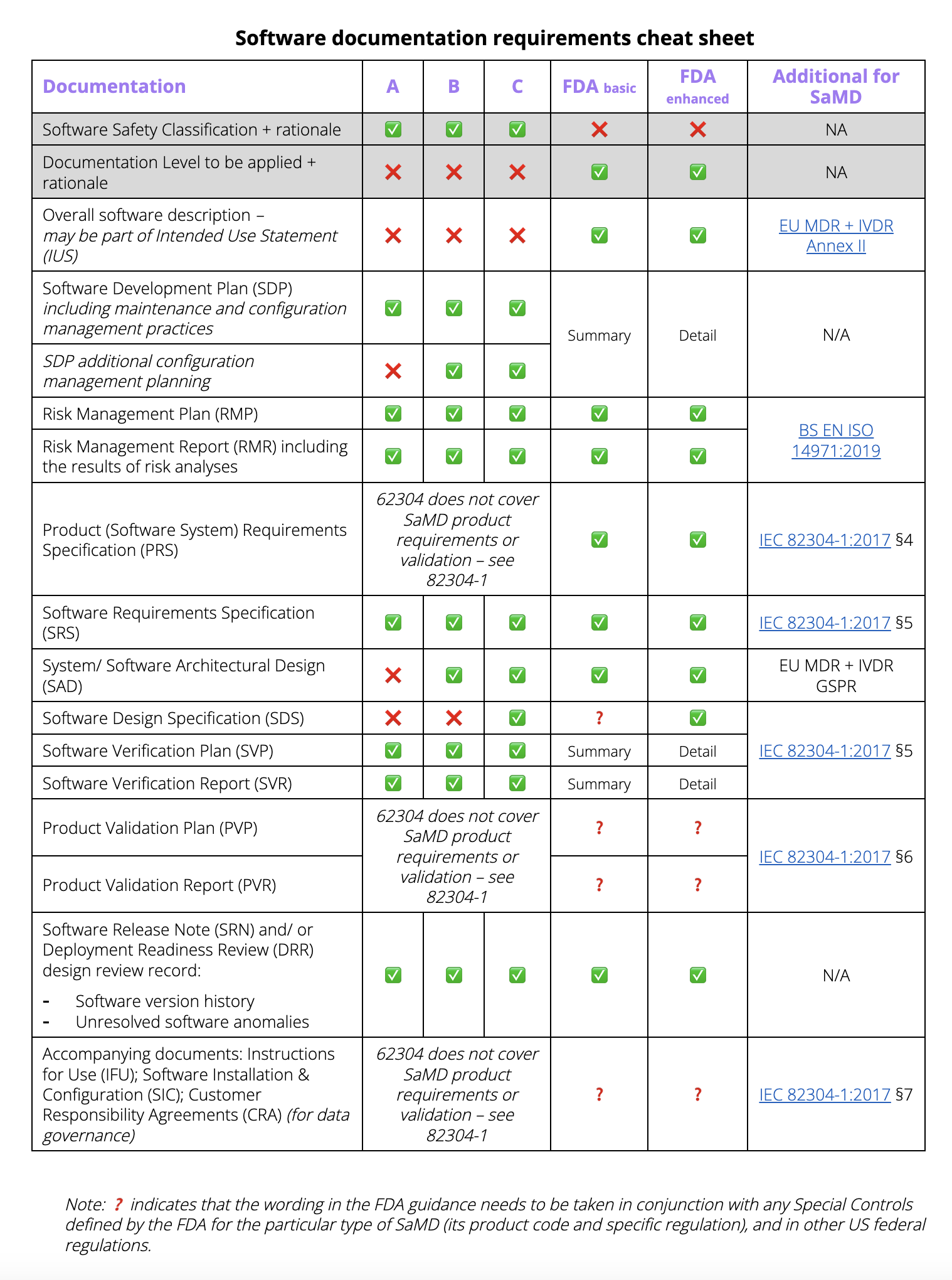
Cheat sheet
Use our free Technical File cheat sheet to get started
Get in touch
Get in touch to see how Hardian can help you create a solid Medical Device File (MDF) Design